Genetic Drift in a Finite Population as an Example of Stochastic Modeling
Jose Antonio Urban Aragon, Ethan Zhong
3/13/2022
Last updated: 2022-03-18
Checks: 6 1
Knit directory:
~/Documents/Winter_Quarter_2022/Fundamentals/jaurbanChicago.github.io/bin/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(12345) was run prior to running the
code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 571343e. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Untracked files:
Untracked: .DS_Store
Untracked: .Rhistory
Unstaged changes:
Deleted: New Folder With Items/.DS_Store
Deleted: New Folder With Items/Genetic_Drift_Markov_Chain.Rmd
Deleted: New Folder With Items/_site.yml
Deleted: New Folder With Items/include/footer.html
Modified: bin/Genetic_Drift_Intro.Rmd
Modified: bin/Genetic_Drift_Markov.Rmd
Deleted: index.html
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (bin/Genetic_Drift_Intro.Rmd) and
HTML (docs/Genetic_Drift_Intro.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 9750a6a | jaurbanChicago | 2022-03-18 | Updated intro |
| html | 9750a6a | jaurbanChicago | 2022-03-18 | Updated intro |
| Rmd | a232a33 | jaurbanChicago | 2022-03-18 | Updated intro |
| html | a232a33 | jaurbanChicago | 2022-03-18 | Updated intro |
| html | decf7fd | jaurbanChicago | 2022-03-18 | Updated intro html |
| html | 50fe36b | jaurbanChicago | 2022-03-17 | Added intro html |
| html | a311740 | jaurbanChicago | 2022-03-17 | Removed all htmls |
| html | d15ed0e | jaurbanChicago | 2022-03-17 | Added intro html |
| html | f8fceeb | jaurbanChicago | 2022-03-17 | Added new intro html |
| Rmd | 049c8d6 | jaurbanChicago | 2022-03-17 | Added intro with new name |
Pre-Requisites
- Wright-Fisher Model
- Discrete Markov Chains
- Basic notions of probability theory
- Basic notions of population genetics
Genetic Drift in Finite Populations
Much population genetic theory concentrates on describing the changes of allele frequencies through time. The two most important factors influencing allele trajectories through time are natural selection and genetic drift [1]. In these vignettes, we will focus on genetic drift and how this process may be modeled as a stochastic process in a Wright-Fisher population.
Genetic drift is the random change of allele frequencies through time in populations of finite size [1,2]. Different random processes could affect the outcome of genetic drift, and they might have nothing to do with the individuals’ fitness [1]. For example, some individuals may die before they reach reproductive age because of extrinsic effects not related to their genetic makeup [1]. Additionally, there is inherent randomness in Mendelian segregation, and these effects could become more apparent when populations are small (we will shortly see why this is the case) [1,2].
Brief Introduction to a Wright-Fisher Model
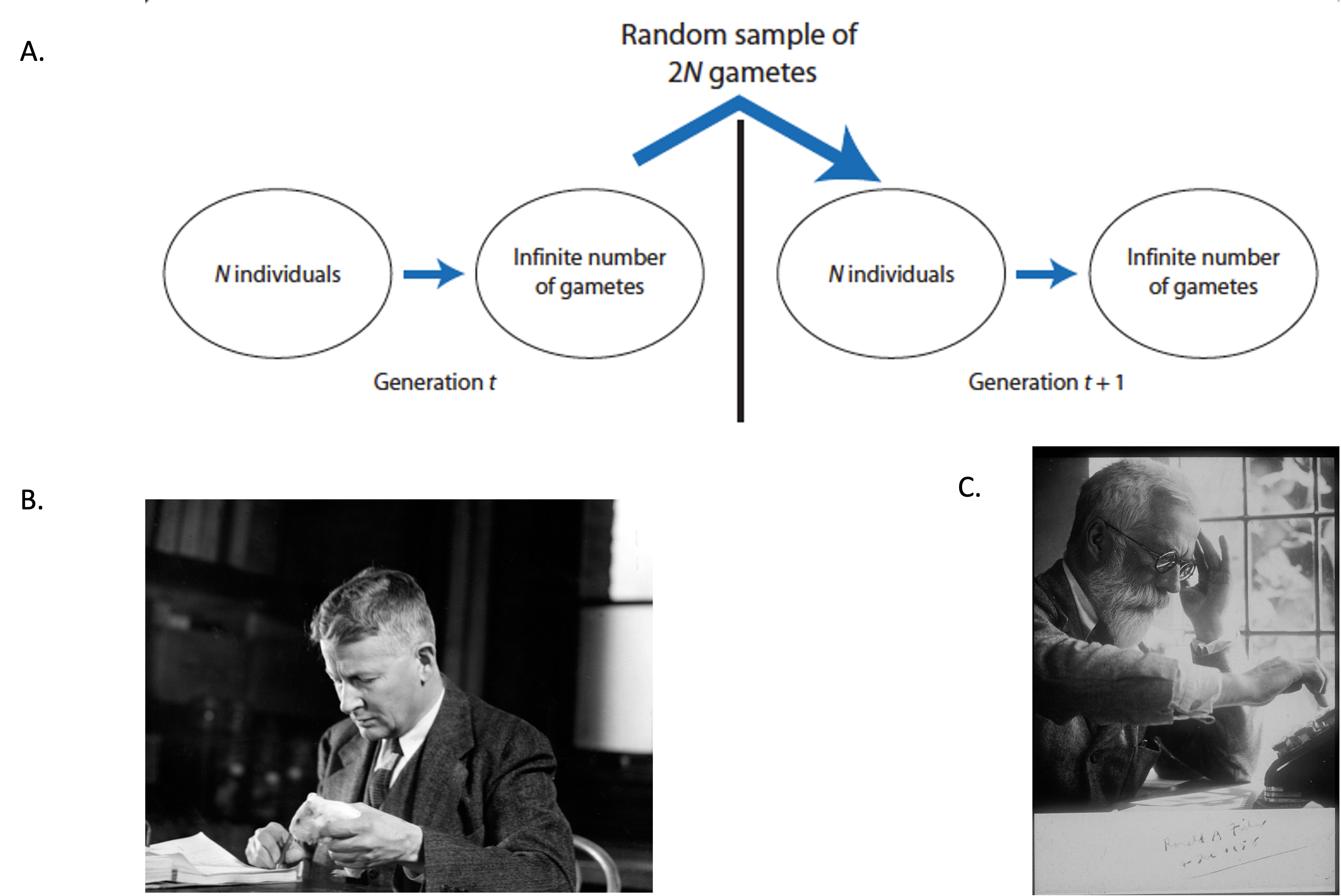
Before delving into the specific probabilistic models of genetic drift, it is important to summarize the main basic assumptions of the Wright-Fisher population. The Wright-Fisher population is an idealized biological scenario where the models of genetic drift are useful to describe allele frequencies changes due to random sampling error [1]. Although the Wright-Fisher model is biologically unrealistic, it has been extremely useful to model genetic drift (and other evolutionary processes) in a relatively simple fashion [1]. Some of the basic assumptions of the Wright-Fisher model include [1]:
- discrete, non-overlapping generations
- numbers of females and males are equal
- size of population (\(N\) individuals ) remains constant through time and is finite
- no natural selection (all individuals are equal in their production of gametes and all gametes are equally viable)
- infinite pool of gametes
- finite sample of \(2N\) alleles is drawn from the gamete pool to form the next generation of \(N\) individuals
- the sampling errors that originate genetic drift occur when the \(2N\) alleles are drawn from the population´s gamete pool
Models Used to Describe Genetic Drift (No Mutation and/or Selection)
In this section of the vignette, we will introduce three basic models that can be used to analyze properties of genetic drift:
- Binomial distribution
- Markov chain
- Diffusion process (continuous time approximation to the Markov chain)
Binomial Distribution
To understand genetic drift of a single two-allele locus in a
Wright-Fisher population, we must first get into the details of an
important discrete probability distribution: the binomial
distribution.
A binomial distribution is a discrete probability distribution that
defines the probability distribution for the sum of \(N\) independent samples of a Bernoulli
variable [1]. Suppose in a diploid population of \(N\) individuals, we have two alleles: \(A\) and \(a\). To describe the sampling of alleles
from the gamete pool of this population, we model the number of \(A\) allele, \(N_A\):
\[ P(N_A = i)={2N \choose i}p^{i}(1-p)^{2N-i} \\ \] This formula describes the probability of sampling \(i\) \(A\) alleles in a sample of \(2N\) alleles from a population where the \(A\) allele has a frequency of \(p\) and the alternate \(a\) allele has a frequency of \(1-p\). The \(p^i\) and \((1-p)^{2N-i}\) terms estimate the probability of observing a \(i\) and \(2N-i\) independent events each with probability \(p\) and \(1-p\), respectively [1]. With some knowledge of the properties of the binomial probability distribution, we can realize that the Wright-Fisher model is a very good approximation to model genetic drift [1]. Some of the properties of the binomial random variables will helps to better understand some specific behaviors of allele frequencies under the action of genetic drift. Let \(p_t\) be the allele frequency of \(A\) at generation \(t\) and \(X_t\) be the number of \(A\) allele at generation \(t\). For a diploid population of constant size \(N\), we have:
\[ \begin{aligned} \mathbb{E}[p_{t+1} \mid p_t, N] &= \mathbb{E}[\frac{X_{t+1}}{2N} \mid p_t, N] \\ &= \frac{1}{2N} \mathbb{E}[X_{t+1} \mid p_t, N] \\ &= \frac{1}{2N}2Np_t \\ &= p_t\\ \\ \text{Var}[p_{t+1} \mid p_t, N] &= \text{Var}[\frac{X_{t+1}}{2N} \mid p_t, N] \\ &= \frac{1}{4N^2} \text{Var}[X_{t+1} \mid p_t, N] \\ &= \frac{1}{4N^2}2Np_t(1 - p_t) \\ &= \frac{p_t(1 - p_t)}{2N} \end{aligned} \] As we may infer from the formulas, the maximum variability in the outcomes of the alleles’ binomial sampling process will occur when the frequency \(p\) of \(A\) equals the frequency \(1-p\) of \(A\) (\(p=1-p=0.5\))[1]. This makes intuitive sense because as the frequency of one of the alleles reaches fixation , the sampling error that results in greatly increased frequencies of the low-frequency allele in one generation will be very highly unlikely [1]. On the other hand, when both alleles are equally frequent or almost equally frequent, sampling error is equally likely to increase or decrease allele frequency and its outcomes could vary more widely along the spectrum of possible allele frequencies.
The following code will demonstrate how the variability of the outcomes of the sampling process at time \(t+1\) depends,in part, to allele frequencies at time \(t\):
library(tidyverse)
# Vector of allele frequency for allele A
p <- seq(0,1,by=0.01) # Frequency p of A allele
N <- 100 # Population of N individuals
# vector to store standard errors
fun_p <- function(p,N){ # function to estimate the standard errors for each frequencies
se <- c()
for(i in 1:length(p)){
se[i] <- sqrt((p[i]*(1-p[i]))/(2*N))
}
return(se)
}
SE <- fun_p(p=p,N=N)
df <- cbind(p,SE)
df <- as.data.frame(df,col.names=names("p","SE"))
# Plot of Allele Frequency p vs Standard Error
ggplot(df,aes(x=p,y=SE))+geom_line()+xlab("Allele Frequency p")+ylab("SE")+ggtitle("Allele Frequency vs Standard Error")+theme(plot.title = element_text(hjust = 0.5))+geom_hline(yintercept = max(SE),color="red")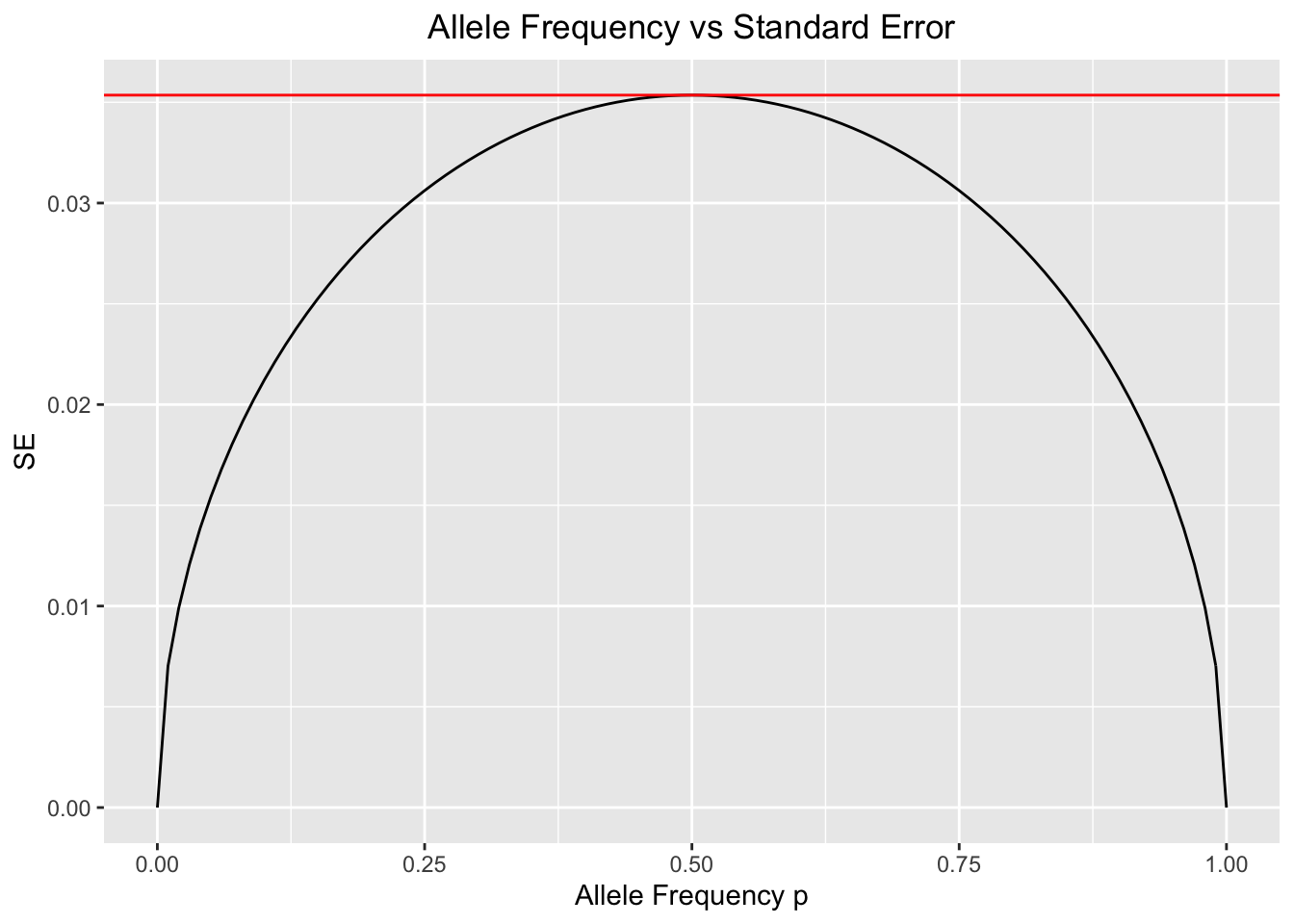
| Version | Author | Date |
|---|---|---|
| 32d40e6 | jaurbanChicago | 2022-03-17 |
#The horizontal red line determines the maximum value of SE.The above plot confirms what we just discussed. The maximum variability of allele frequency changes happens when both alleles are very close in frequency[1].
Another feature affecting the variability of binomial sampling errors in genetic drift is the population size. As population size increases, the variability of allele frequencies becomes less and less, constraining the spectrum of possible changes in allele frequencies moving forward in time and reducing the variance of the sampling error process [1,2].
The following code will illustrate this:
p <- 0.5 # Set the p frequency to 0.5
N <-seq(10,100,by=10) # Create the N vector
fun_p <- function(p,N){ # function to estimate the standard errors for each population size
se <- c()
for(i in 1:length(N)){
se[i] <- sqrt((p*(1-p))/(2*N[i]))
}
return(se)
}
SE <- fun_p(p=p,N=N)
df <- cbind(N, SE)
df <- as.data.frame(df,col.names=names("N","SE"))
# Plot of Allele Frequency p vs Standard Error
ggplot(df,aes(x=N,y=SE))+geom_line()+xlab("Population Size (N)")+ylab("SE at p=0.5")+ggtitle("Population Size vs Standard Error at p=0.5")+theme(plot.title = element_text(hjust = 0.5))+geom_hline(yintercept = max(SE),color="red")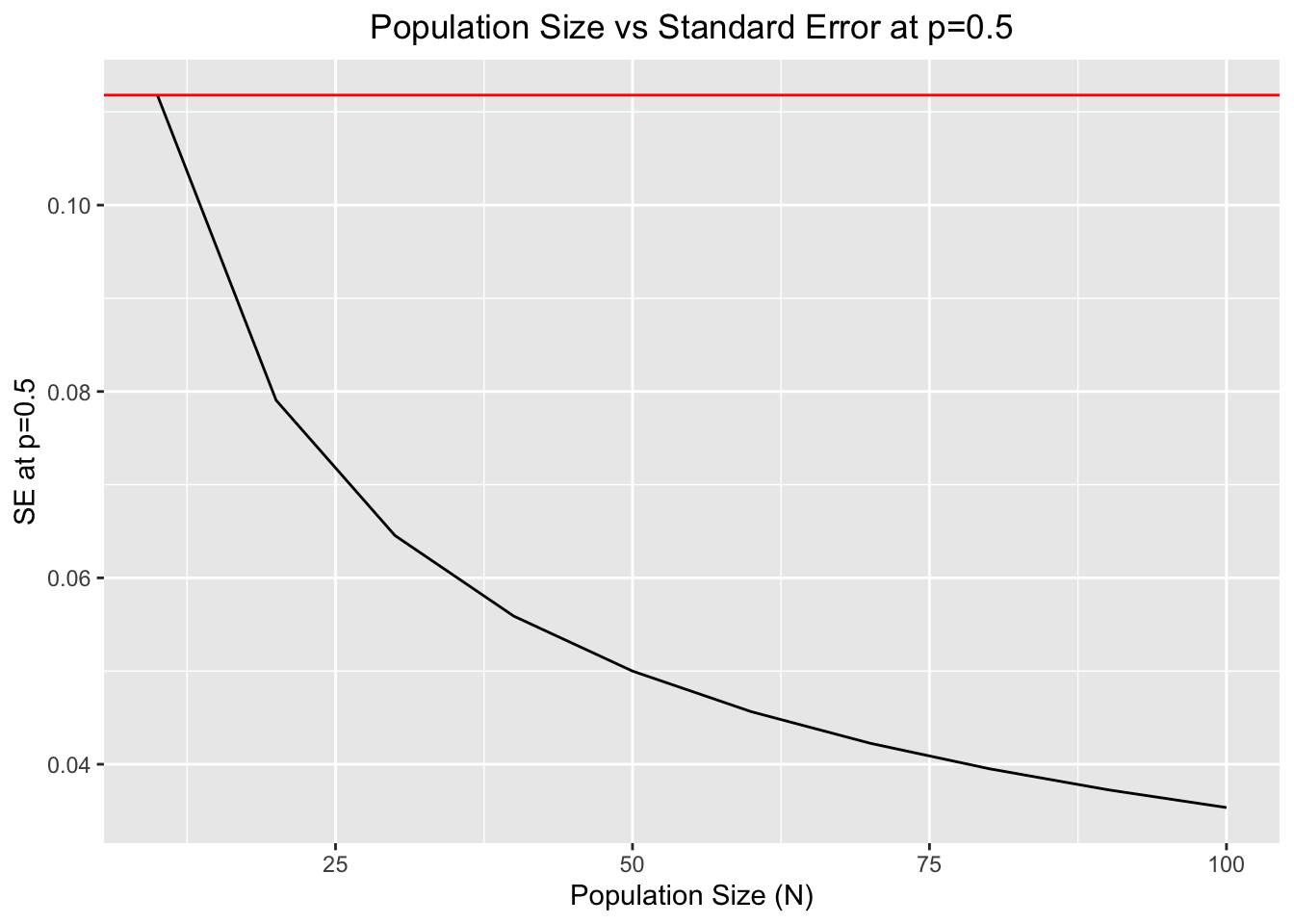
| Version | Author | Date |
|---|---|---|
| 32d40e6 | jaurbanChicago | 2022-03-17 |
#The horizontal red line determines the maximum value of SE at p=0.5.Both features (population size and allele frequencies at time \(t\)) will be of paramount importance to determine the outcomes of genetic drift in a given Wright-Fisher population.
Markov Chain
The Markov chain model of genetic drift will be explained in the following vignette.
Diffusion Process (Continuous time approximation of the Markov chain model)
The diffusion process model of genetic drift will be explained in the following vignettes:
References
Hamilton, M. B. (2008). Population genetics. John Wiley & Sons, 53-73.
Hartl, D. L. (2020).A primer of population genetics and genomics. Oxford University Press,147-173.
sessionInfo()R version 4.1.2 (2021-11-01)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Catalina 10.15.7
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] forcats_0.5.1 stringr_1.4.0 dplyr_1.0.8 purrr_0.3.4
[5] readr_2.1.2 tidyr_1.2.0 tibble_3.1.6 ggplot2_3.3.5
[9] tidyverse_1.3.1
loaded via a namespace (and not attached):
[1] Rcpp_1.0.8.2 lubridate_1.8.0 assertthat_0.2.1 rprojroot_2.0.2
[5] digest_0.6.29 utf8_1.2.2 R6_2.5.1 cellranger_1.1.0
[9] backports_1.4.1 reprex_2.0.1 evaluate_0.14 highr_0.9
[13] httr_1.4.2 pillar_1.7.0 rlang_1.0.2 readxl_1.3.1
[17] rstudioapi_0.13 whisker_0.4 jquerylib_0.1.4 rmarkdown_2.11
[21] labeling_0.4.2 munsell_0.5.0 broom_0.7.12 compiler_4.1.2
[25] httpuv_1.6.5 modelr_0.1.8 xfun_0.29 pkgconfig_2.0.3
[29] htmltools_0.5.2 tidyselect_1.1.2 workflowr_1.7.0 fansi_1.0.2
[33] crayon_1.5.0 tzdb_0.2.0 dbplyr_2.1.1 withr_2.5.0
[37] later_1.3.0 grid_4.1.2 jsonlite_1.8.0 gtable_0.3.0
[41] lifecycle_1.0.1 DBI_1.1.2 git2r_0.29.0 magrittr_2.0.2
[45] scales_1.1.1 cli_3.2.0 stringi_1.7.6 farver_2.1.0
[49] fs_1.5.2 promises_1.2.0.1 xml2_1.3.3 bslib_0.3.1
[53] ellipsis_0.3.2 generics_0.1.2 vctrs_0.3.8 tools_4.1.2
[57] glue_1.6.2 hms_1.1.1 fastmap_1.1.0 yaml_2.2.2
[61] colorspace_2.0-3 rvest_1.0.2 knitr_1.37 haven_2.4.3
[65] sass_0.4.0 This site was created with R Markdown